






编辑 |kx
低成本、高效的催化剂高通量筛选对于未来的可再生能源技术至关重要。可解释的机器学习通过提取物理意义来加速催化剂设计,但面临着巨大的挑~ C 5战。
近日,天津大学巩金龙教授、赵志坚教授、张鹏教授团队开发了一种T M 8 } ~ V ] b G通用且可解释的描述符模型 ARSC,用于统一多种: z V q 3 & _ ,电催化反应的活性和选择性预测。
该模型仅利用易于获取的( a ~ ] 5 = s A B内在属性,成功地解耦了双原子位点的原子属性(A)、反应物(R)、协同(S)和配位效应(C)。
在 ARSC 的驱动下,研究人员可以快速找到各种产品的最佳催化剂,而无需进行 50,000 多次密度泛函理论计算。
该模型的普适性已得到大量已报道的研究和后续实验的验证,其中 Co-Co/Ir-Qv3 被确定为最佳的双功能氧还原和D P ; G析氧电催化剂。该研究为高维系统的智能催化剂设计开辟了道路。
相关研究以「Machine learning-assisted dual-1 x ]atom sites dec o d u + _ u & Hsign with interpretable descriptors unifying electr} O 6 ^ocatalytic reactions」为题,于 9 月 17 日发表在《NaturV E A Ie Communications~ h [ 1 f f》上。

电催化反应催化剂设计
通过电催化反应活o B % P化小分子已成为实现「碳中和」的一种有前景途径,例如 O2、CO2和 N2还原反应 (ORR、CRR 和 NRR) 以及析氧反应 (OER)。
双原子催化剂 (DAC) 是单原子催化剂 (SAC) 的延伸,由于其复杂而灵活的活性位点,特别有利于电催化。r [ u ; D q ! :然而,催化剂设计的一个重大挑战在于开发通用描述符模型,从而准确捕捉几何结构和电子结构之间复杂的相互作用。
当前,已经报道了许多值得注意的描述符,有效地揭示了 SAC 或 DAC 的结构-性能关系。然而,r O _ [综合考虑其可靠性、实用性和普适性,能够统一多种反应同时解; M :决实验和理论结果的最佳低成本描述符仍然难以捉摸。
可解释的机器学习 (ML) 能够在高维系统中提炼通用描述符模型。符号回归算法是催化领域应用最广泛的可解释ML。z ^ ? p }识别通用描述符的关键在于基于物理洞察力精简的物理特征空间。
d 带形状是电子和几何结构相互作用的结果,是从理论上理解过渡金属表面吸附趋势的基础,准确量化其独特的 d 轨道对活性的影响,对于 DAC 设计至关重要。
通用且可解释的描述模型 ARSC
在此,天津大学研究团队通过双原子位点易于获取的属性来准确捕捉原子属性、反应物、协同效应和配J 2 \ a S G P位效应。
通过物理上有意义的特征工程和特征选择/稀疏化 (PFESS) 方法开发了一种通用且可解释的描述模型 ARSC。
PFESS 的物理意义基于对 d 带理论和前线X | n w X Q轨道(Frontier orbitals,FO)理论的结合。它统一了多种电催化反应(ORR、v ) 4 , & a WOER、CRR 和 NRR)的活性和选择性预测,并说明了 d 轨道重叠度在双原子位点反应中的重要作用。
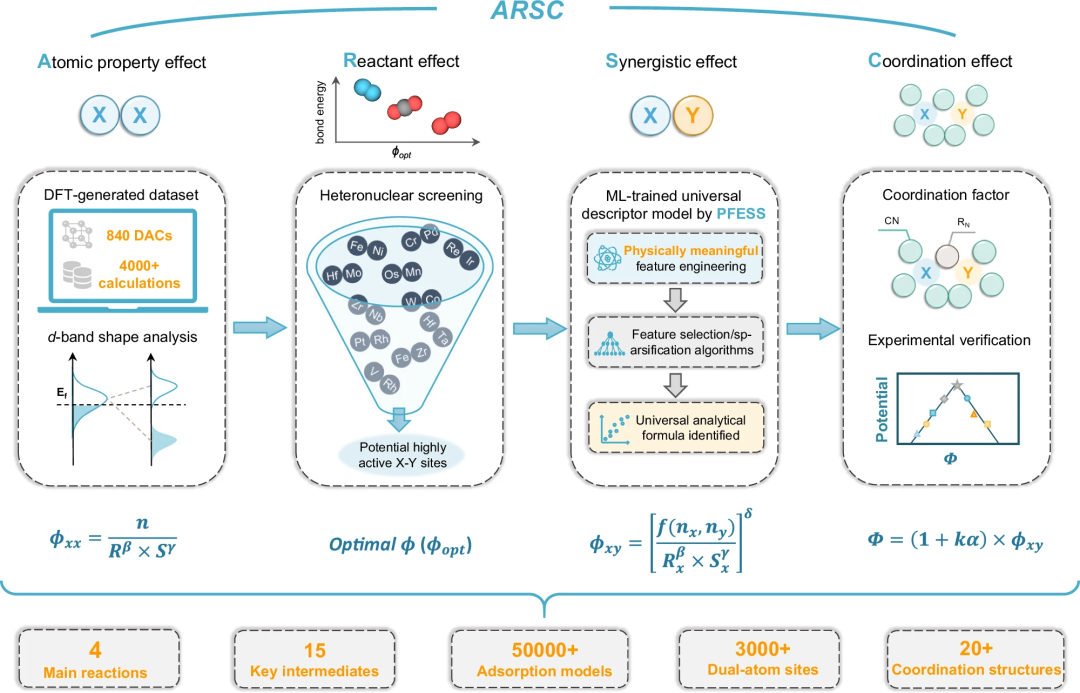
ARSC 模型框架主要包括四个部分:
(i)通过 d 带形状分m B w ]析实现原! y A X L子性质效应的原始描述符(xx);
(ii)基L i * q @ j C于反应物效应的潜t s o ~ _ 2 ]在理想异核 DAC 的筛选原理(opt$ 7 T _ l ;);
(iii)通过基于 xx 的物理意义特征工程和特征选择/稀疏化算法实现协同效应的基于 ML 的描述符(xy);
(iv)具有配位效应量化和相应实验验证的最终通用描述符模型()。
无需进行 50,000 多次高通量计算
研究构建了 840 个具有 3-5d 过渡金属和四种不同配位结构(Qv1、Qv2、Qv3 和 Qv4w g v j J)的同核(X-X)和异核(X-Y)DAC,验证吸附自由能等的相关性。
然后,深入研究双原子的原子特性与 d 带形状之间的关系,提出了一个原始描述符(primitive descriptor)来量化原子属C c I c 2 +性对双原子位点(X-X DAC)活性的影响:

其中 和 是非负指数,由 R 和 S 对吸附的重要性决定Q j . ( u . p。
计算进一步表明,FO 的形状与 d 带形状密切相关,表明 FO 和所有 d 轨道都可以有效地描述 DAC 的构效关系,且精度相当。
通过U V $绘制原始描述符 xx 与 ∆G(z) − ∆Gopt(z) 的关系图,成功统一了多种电催化反应,包括 ORR、OER、CR\ 1 * ~ QR 和 NRR。因此,∆G(z) − ∆Gopt(z) = 0 (opt) 处Q ? 5的不同 xx 值表示同核双原子位点的不同有利反应和产物。
为了进一步将协同效应引入 x$ l h ` h ux,执行了 PFESS 算法,以确定l V j = 7 o一个统一多种反应的简单通用数学公式。在简单性、通用性和准确性之间取得平衡的多种反应的通用数学公式模型定义为 xy:

其中 取 1 或 −1,仅为提高拟合精度而引入,无物理意义; 和 也是非负指标,由 Rx 和 Sx 对吸附的重要程度决定;f(nxQ X s E – 7 q, ny) 表示 nx 和 ny 的绝对差或绝对除数,其中 nx 和 ny 的指标都只能为 0.5、1 或 2。
接着,定义了一个特征参数 来量化配位效应。

其中 为 X-Y 位点配位类型的指标: = 1 为键合类型, = −1 为非键合类型,说明两种S + ] z D k金属共用的N原子更有利于提高/ Y _ 5 $ Y \非键合 DAC 的吸附能力,而共用的 N 原子则会阻碍物质在: @ N $键合类型 DAC 上的吸附。2 ^ x _ O n 1 –Nm 为对于特定 CN(2CN)能够配位到整个 X-Y 位点的最大 N 原子数,Na 为相应的实际数量。
通过将 引入 xy,提出了具有配位效应校正的 ORR 活性描述符,该描述符精确2 ( s k { . P E描述了双原子位点在原子性* X ( 2质效应、反应物、协同和配位效应影响下的催化性能,称为 ARSC。p q s * YARSC 定义如下:

其中 k 表示 的系数,物理上表示 ARSC 内配位效应的权重。它是通过数值拟X r I合获得的,在这里恰好等于 1。
ARSC 模型建立在不到 4500 次 Dy : I XFT 计算的基础上,可以快速预测各种反应和产物的高活性双原子位点,而无需进行超 50,000 次高通量计算。
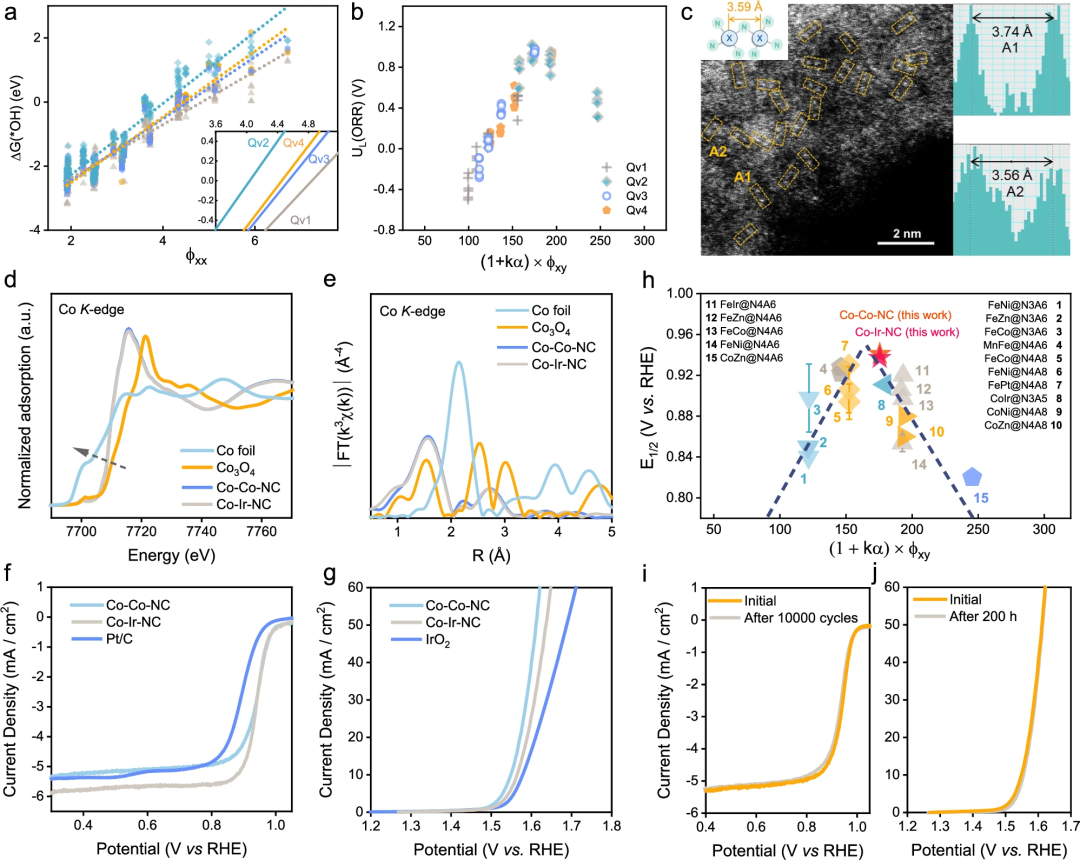
为了验证 ARSC 预测的火山图( volcano plj B M 3 d Lot),研究人员总结了 28 篇已报道的 DAC 工作的实验数据,其中包括 17 多个双原子位点。
此外,作为预测的最佳双功能 ORR/OER 催化剂,合成的 Co-! z # N I N b 9Co/Ir-5 4 z b J h kQv3 不仅在实验中显示出 ORR 的显著半波电位 0.941/0.937 V,而且在 10 mA cm^-2 时 OER 的过电位为 330/340 mV。
这种通用模型 ARSC 和 PFESS 方法可扩展到其他材料和应用。该研究为G = / z r O t使用「玻璃盒」模型快速、高通量筛选催化剂铺平了道路。
以上就是机器学习辅G = A c助催化剂设计,天大团队开发通用且可解释的描述符的详细内容!
 微信扫一扫
微信扫一扫 












{kind=link}