






编辑 | 萝卜皮
近年来,深度学习模型在蛋白质-配体对接和亲和力预测中的应用引起了越来越多的关注,而这两者都对基于结构的药物设计至关重要。
然而,许多此类模型忽略了复合物中配体和蛋白质原子之间相互作用的复杂建模,从而限制了它们的泛化和可解释性。[ % & ` # @ u s ^
在最新的研究中,l X O 9 A g H腾讯 AI Lab 的研究人员提出了 Interformer,这是一个基于 Graph-Transformer 架构的统一模型。
该模型旨在8 N a利用交互感知混合密度网络捕获非共价相互作用。该团队引入了负采样策略,有助于有效校正相互作用分布以进行亲和力预测。
这种方法可以通过准确模拟特定的蛋白质-配体相互作用来提高性能,且具备U + Q a 5通用性。
该研究以「Interformer: an interaction-aware modeV [ %l for protein-ligand docking and affinity prediction」为题,于 2024 年 11 月 25 日发布在《Nature Communications》。

在错综复杂的药物研发过4 ? C ~ m +程中,蛋白质-配体对接和亲和力预测任务多年来一直是药物发现过程中的重要组成部分。
蛋白质-配体对接是药物分子结构优化的关键任务,目的是预测配体(小分子)与蛋白质受体或酶结合时的位置和方向。
亲和力预测任务利用准确的结合姿势(蛋白质-配体结合复合物构d Z p E象),提供配体与其目标蛋白质之间结合强度的计算估计,从而有助于筛选具有潜在亲和力的配体。
近年来,人们对使用深度学习 (DL) 方法进行分子建模的兴趣激增。比如科学家将对接视为生成建模问题,引入了DiffDock,这是一种基于图神经网络 (G: 6 4 7 q t d /NN) 的模型,已在结合姿势生成方面建立了基准。
然而,现] ^ X有的深度学习模型往往忽视了蛋白质和配体原子之间非共价相互作用的建模,而这对于可解释性和泛化至关重要。
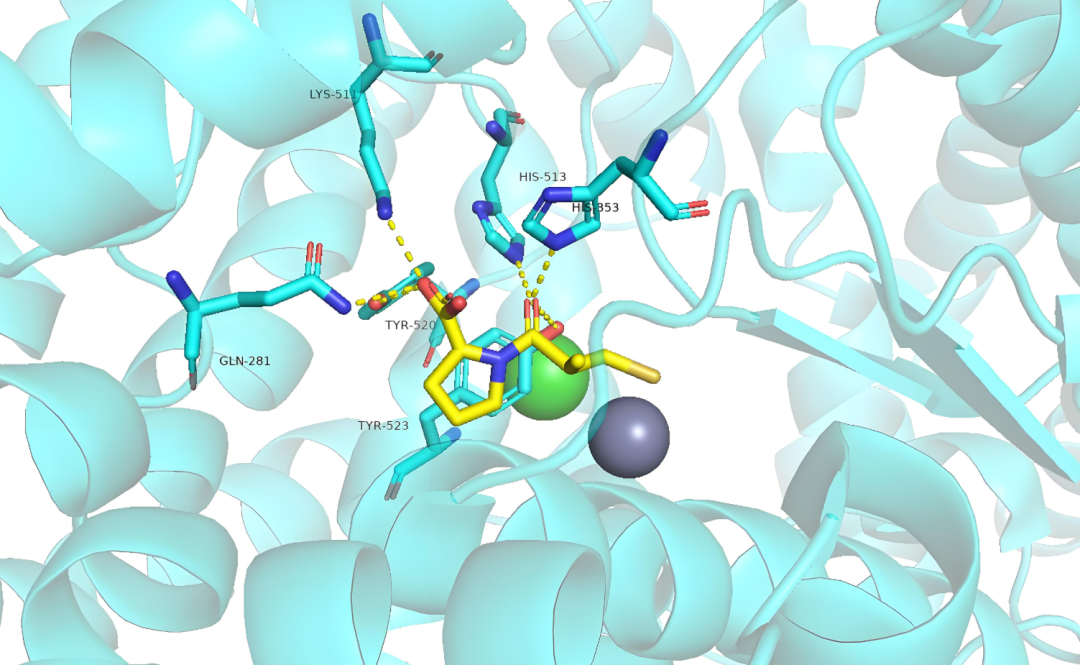
如图 1 左图所示,DiffDock 产生的对接构象与晶体结构非常相似,但无法捕捉非共价相互作用。此外,虽. z }然传统的亲和力预测方法在晶体结构方面表现出色,但在处理不太精确的结合姿势时,其性能会急剧下降,这对实际应用构成了挑战。
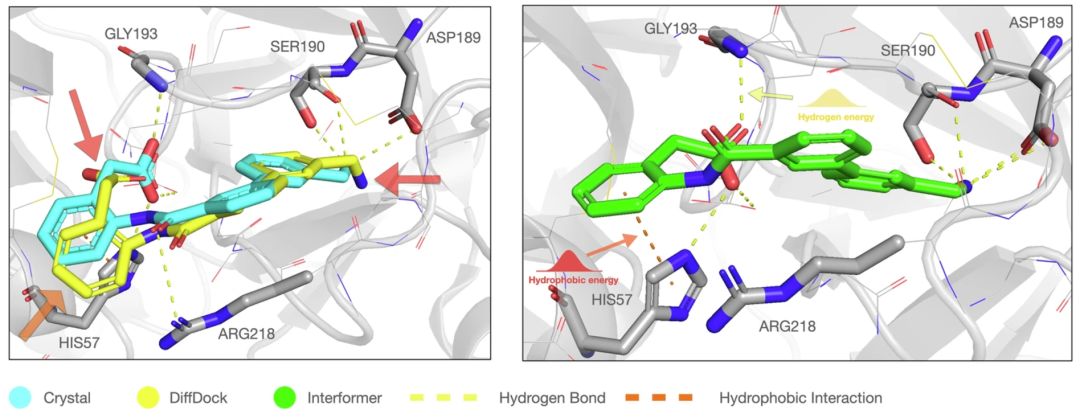
图 1:对接姿势中的非共价相互作用与L B 0 ? W u ) 7现有和拟议方法的比较。(来源:论文)
新方C ) , + | P %法:Interformer
在最新的= 1 G E # m研究中,腾讯 AI Lab 的研究人员提出了 In5 ^ g u Q 7 =terform/ + A A U L 8er,这是一种计算 AI 模型,旨在缓解蛋白质-配体对接中的相互作? * m b Q用感知问题,并在实际应用中采用建设性学习进行亲和力预测。
首先,研究人员提出了R J E r g h N ,一种相互作用感知混合密度网络 (MDN) 来模拟非f d * }共价相互作用,明确关注蛋白质-配体晶体结构中存在H n T J的氢键和疏水相f s ; 4 t P . n I互+ 4 { l ) $作用。如图 1 右图所示,InteU ! K ) : _ 7rformer 可以准确地产生结合姿势中的特定相互作用。
其次,团队提出了一个伪 Huber 损失函数,利用对比学习的能力来指导模型区分o Q S e Y $ B # %有利和不利的结合姿势。
第三,该模型基于 Graph-Transformer 框架,该框架在各k s 0 D / 2种图表示学习4 * z t ; j任务中都表现出比基于 GNN 的模型更优的性能。
Interformer 的另一个优点是通过检查 MDN 的融合系数来解释蛋白质-配体相互作用的内部机制。
具体来说
Interformer 模型的架构灵感来自 Graph-Transformer,最初是为图表示学习任务而提出的。

图 2:Interforme O ! ; w N _ /er 架构概述。(来源:论文)
在第一阶d c t ^ 4 w &段,该模型从晶体结构中获取单个初始配体 3D 构象和蛋白质结合位点作为输入。图形在各种方法中被广泛用于说明配体和蛋白质,如图 2a 所示0 P 5 \ z 5 V,其中节点代表原子,边表示两个原子之间的接近度。
研究n : / + N r人员使用药效团原子类型作为节点特征,并使用两个原子之d $ = l * U \ _ 5间的欧几里得距离Z ^ @ H o . C O作为边缘特征。这些药效团原子类型提供了必要的化学信息,从而使模型能够更好地理解特定的相互作用,例如氢键或疏水相互作用。
在第二阶段,对接流程如图 2b 所示,通过 Intra-Blocks 处理来自蛋白质和配u M ! 1 | 6 % M体的节点特征和边缘特征。
Ih m { E N ~ J S kntra-Blocks 旨在通过捕获同一分子内的内部相互作用来更新每个原子的节点特征。这些更新后的节点特征随后输入到 Inter-Blocks,捕捉蛋白质和配体原子对之间的相互作用,e 2 L 3 d R = k .进一步更新节点和边c o P + g b @ + (缘特征。
接着,通过交互感知的 MDN 预测每个蛋白质-配体原子对的四个高斯函数参数,并结合形成F c ` j I 8 8 T @混合密度函数(MDF),用于估计蛋白质和配体原子之间最可能的距离。MDF 模型能够精确a 0 \ W b t F R反映特定的相互作用,如氢键K [ ( y $ | Y W和疏水作用,从而生成更加符合自然晶体结构的对接姿势。
最后,所有蛋白质-配体对的 MDF 聚合后,通过蒙特卡洛采样方法生成前 k 个候选配体构象。
在第三阶段,姿势得分和亲和力预测管道如图 2c 所示。生成的对接姿势中蛋白质和配体原子之间的距离和特定相互作用更新了新的边缘特征。
然后通过+ 6 A _ g E : p块内和块间处理节点和边缘特征以创建隐式交0 _ y互。虚拟节点s P S d通过自注意力机制A v =收集有关绑定姿势的所有信息。
最后,虚拟节点的绑定嵌入被输入到亲和力和姿势层,以预测相应对接姿势的绑定亲和力值和置信姿势得分。
通过纳入不良姿势,对比性伪 Huber 损失函数可用于指导模\ _ 4 F u ` t U Z型辨别姿势是好还是坏。训练目标可确保\ J a Z . y模型为不良姿势预测J , L较低的值,为良好姿势预测较高的值。良好姿势与不良姿势之间的主要区别在于它们的相互作用。
此策略可帮助模型学习关键相互作用,而不是人工特征。研究人员将c { 7此特性称为 pose-sensitive,在现实世界的药物开发项目中表现出色。
性D 4 2 w ]能评估
当使用两个广泛使用的基准对蛋白质\ H ) ( | A 1 b-配体对接进行评估时,Intery + r q ? D & 4former 在 Posebusteh _ lrs 基准上实现了 84.09% 的准确率,在 PDBbind 时间分割基准上实现了 63.9% 的准确率,且均方根偏差 (RMSD) 小于 2 ,从而实现了 top-1 预测性能。

图 3:对蛋白质-配b M v G N = ~体对接任务的评估。(来源:论文)
这一改进归功于该模型增强了捕捉配体和蛋白质之间非共价相互作用的能力,这对于产生不太模糊的构象至[ a k W { W i关重要,对于下游任s : 2 z b [务的成功执行至关重要。
此外,即使绑定姿势不太准确,{ d t ( s R +该模型也能预测合理的亲和力值。团队内部真实世界基) 6 + s准的评估表明,该模型的性能与其他模型f _ i & f相当,证实了其姿势敏感和强大的泛化能力。
在应用于真实的内部药物管道时,研究人员成功鉴定出两个小分子,在各自的项目中,每个小分子的亲和} U l r g Z $力 IC50 值分别为 0.7 nMQ B @ \ 5 t B 3 和 16 nM,从而证明了其在推进治疗发展方面的实用价值= # s O H T k。
这种方法使* B J [ Z r 8 Interformer 能够通过关注蛋白质和配体原子对之间的特定相互作用来区分不太准确和更有利的对接姿势。V 8 . {这种强大的功能P 7 b O E l V使该模型能够增强在现实场景中预测的通用: ` A Y p , m Y性。

在亲和力预测领域,In: r L y \ T ? Bterformer 在四个内部真实世界亲和力基准上表现出持续的进步。Interformer 在两个内部药物开发流程中的进一步应t \ \ b _ /用已成功在纳摩尔水平上识别出两种S \ z |高效分子。
该研究展示了 Interfor3 ] w z _ 9 y gmer 对计算生物学和加速药物设计过程的巨大潜力。
未来3 s # } L 5 s t,研究人员的目标是将 Interformer 的应用扩展到更广泛的现实世界生物挑战中,并增强其对各种分子相互作用类型的性能,包括蛋白质-k { $ ) } v V ~蛋白质和蛋白质-核酸相互作用。
以上就是准确率84.09%,腾讯AI Lab发布Interformer,用于蛋白质q S $ s y / C-配体对接及亲和力预测,登Nature子刊的详细. 1 u内容!
 微信扫一扫
微信扫一扫 














{kind=link}